
West Nile Virus (WNV) is an arthropod-borne virus of the family Flaviviridae, genus Flavivirus—the same family as other significant pathogens, including Zika and yellow fever [1, 2]. It is further characterized by different genetic lineages, where the most important considerations in human and animal health are Lineage 1a and Lineage 2. Both lineages were detected in the MENA region, with Lineage 1a traditionally having a wider distribution, including Africa, Europe, the Middle East, Asia, and the Americas [3, 4]. On the other hand, Lineage 2 has recently emerged as a dominant strain associated with much more severe outbreaks and higher mortality rates in humans and animals [4]. The first isolation of WNV was in the West Nile District of Uganda in 1937; however, in the Middle East and North Africa (MENA) region, the first confirmed outbreak was reported in Egypt in1950, involving a febrile illness with neurological complications that primarily affected children. Subsequently, the first outbreak of neuroinvasive disease in elderlies caused by WNV was reported in Israel in 1957 [5]. The virus is transmitted mainly through mosquito bites with Culex species, especially Culex pipiens, serving as the primary vector in most regions [4, 6]. WNV affects a wide range of species, with birds serving as the main reservoir, and humans or horses—who act as incidental hosts—are considered dead-end carriers, incapable of developing viral entities for transmission [7]. According to the Centers for Disease Control and Prevention (CDC), approximately 80% of human WNV infections are asymptomatic [8].
In comparison, about 20% develop mild symptoms, often described as West Nile fever—characterized by fever, headache, fatigue, and sometimes nausea. However, less than 1% of infected individuals experience severe neuroinvasive diseases such as meningitis, encephalitis, or acute flaccid paralysis, particularly in the elderly and immunocompromised individuals [9, 10]. The MENA region's climatic factors, migratory bird pathways, and urbanization have all contributed to the virus’s persistence and periodic outbreaks [11-13]. Over the following decades, sporadic cases of WNV were documented in other parts of the MENA region, including Israel, Algeria, Morocco, and Tunisia, confirming the virus’s broader presence in the area [14].
The epidemiology of West Nile Virus (WNV) has become increasingly concerning in recent years, with a notable resurgence observed across several countries in the Middle East and North Africa (MENA) region, marked by a significant increase in reported cases among humans and animals; for example, in Israel, 2018 saw a significant WNV outbreak with over 136 human cases [6, 15]. In 2016, the Central Veterinary Research Laboratory in Dubai, United Arab Emirates (UAE), reported the first isolation of WNV in a dromedary calf, confirming the virus's presence in the country, where the zoonotic transmission likely occurred through human-animal interactions, particularly involving infected Arabian camels in Saudi Arabia; which suggests that the virus circulates through natural transmission cycles globally [16]. In 2020, many samples from veterinarians and horses were collected in Palestine, revealing the area's notable prevalence of WNV [17]. In 2022, Pakistan experienced a sharp increase in mosquito-borne Flavivirus diseases, such as dengue, for simple reasons, including climate change and inadequate sanitation [18]. However, a key driver behind the resurgence of WNV in the MENA region is environmental changes, including rising temperatures and altered precipitation patterns due to climate change. Warmer temperatures and extended summer seasons create ideal conditions for Culex mosquito populations, allowing them to prolong their active transmission period [12].
Given the growing impact of the West Nile virus in the MENA region, this study aims to analyze its genetic features and transmission patterns to understand its recent spread and evolution better. This is the first phylogenetic study using published WNV sequences from the MENA countries. By comparing older strains with those from recent outbreaks, we aim to explore how the virus has evolved and identify any genetic shifts that may enhance its spread or survival in both nucleotide and amino acid levels through constructing and examining phylogenetic trees, we explore the virus’s evolutionary dynamics and trace infection patterns among various hosts. Our objective is to determine which countries are most affected and assess whether the virus's re-emergence poses a significant public health threat, providing valuable insights to improve the region's WNV surveillance and control efforts.
We collected data using carefully defined parameters to ensure accuracy and reliability. Our primary resource was the NCBI Virus database, known for its high-quality and trustworthy genomic data. We identified the MENA region according to the World Bank’s classification and found 210 WNV sequences from Egypt, Morocco, Tunisia, Iran, Iraq, Israel, Pakistan, and the United Arab Emirates (UAE). The following selection was based on their completeness, collection date, and length: 11 sequences were chosen based on their complete nucleotides, and another 17 partial ones were considered as they exceeded 1000 base pairs (bp) for better informative results. As for host picking, we focused on insects, particularly mosquitoes from the Culex and Culicidae families—being the primary vectors for WNV transmission to humans (Homo sapiens). However, to cover all relevant years and potential relationships, we included sequences from mammals such as horses (Equus caballus), camels (Camelus dromedarius), and mice (Mus musculus), as well as from avian hosts, including members of the Anatidae and Ciconiidae families. Our selection aimed to include a representative sequence for each available year, country, and host, so any ambiguous sequences or erroneous data were replaced with the closest version of the same, similar length or excluded altogether to avoid distorting the results. After applying our criteria, we obtained 26 sequences for further analysis.
Our results were produced using a range of software tools. For the initial multiple-sequence alignment, we used the MAFFT program, which is known for its sensitivity and reliability in handling large datasets accurately. We used the MUSCLE alignment in MEGA 11 for cross-validation and sequence visualization, removing ambiguities. We applied the default gap penalties for MUSCLE alignment, with a Gap Open value of -400.00 and a Gap Extend value of 0.00. We also used the commonly applied UPGMA clustering algorithm throughout the initial and later iterations. Since UPGMA assumes a constant rate of evolution, it was a good fit for our dataset, which spans relatively short periods. Based on the results, additional programs like R Studio and UGENE were used to generate different graphs, as well as a web-based statistical analysis tool and advanced visualization tool [19].
We used two statistical methods for our analysis: Maximum Likelihood (ML) and Neighbor-Joining (NJ). For the ML approach, we ran 100 bootstrap replications to assess and support the reliability of the inferred tree. Given the expected differences in transition and transversion rates in viruses, we selected the Tamura-Nei model, which we saw as the most appropriate choice for our data. We used Gamma distribution for more realistic results accounting for site-to-site rate differences. However, we went with complete deletion for gaps or missing data, keeping only the reliable and aligned sequence's parts, reducing inconsistencies caused by incomplete data, with the Nearest-Neighbor-Interchange (NNI) algorithm as a default heuristic method in ML approaches exploring different tree topologies with minimal computational burden, gradually improving the tree's accuracy.
Additionally, all codon positions were included to ensure that every sequence aspect was analyzed. In the second phylogenetic analysis, we used the Neighbor-Joining (NJ)—a less computationally intensive option—often used when dealing with large datasets for approximate results. We assessed the tree’s stability using 1000 bootstrap replications and the p-distance model, which calculates the proportion of nucleotide differences between sequences. Like the ML method, we applied a Gamma distribution, opted for complete deletion to manage missing data, and included all codon positions for consistency. In the final step of phylogenetic tree interpretation, we applied different rooting methods depending on the dataset. In Figure 1, we used the default midpoint rooting, which does not require an outgroup; this method places the root at the midpoint of the longest path between the two most divergent sequences in an unrooted tree and is particularly suitable for datasets with high coverage values that are suitable for our analysis [20, 21]. Figure 2 also used the default midpoint rooting; however, the tree was manually organized to emphasize lineage relationships and visually highlight key findings. In contrast, for the two phylogenetic trees shown in Figure 3, we used outgroup rooting using reference sequences from Lineage 1a and Lineage 2, assuming that one or more taxa are distinct from the leading group. The branch connecting them serves as the starting point for interpreting evolutionary relationships within the tree, which allows us to demonstrate genetic divergence clearly and to show the evolution pat; this assumes that one or more of the taxa are divergent from the rest of the ingroup and the branch linking the ingroup and outgroup becomes the starting point and defines all subsequent evolutionary events within the tree [20].
Four sequences were chosen to represent the dataset's most extended and most genetically diverse sequences, allowing a comprehensive comparison between the two major lineages and the most recent complete sequence from the MENA region. We used MUSCLE to align the sequences, keeping the same settings as before, and the data were extracted from MEGA for manual analysis. Key amino acid variations were then organized into tables for further discussion, highlighting the evolutionary similarities and differences.
To better understand the circulating WNV in the MENA region, we analyzed the relationships between all the complete sequences using the Maximum Likelihood (ML) method. The phylogenetic tree (Figure 1) reveals two main lineages in the region: Lineage 1a and Lineage 2. Lineage 1a includes WNV strains from Israel (2000, 2002, and 2003) and Tunisia (2003), all clustering with high bootstrap support (100%), indicating strong confidence in their consistent evolutionary relationship. Additionally, strains from the UAE (2015) and Morocco (2005) isolated from camels and horses form a distinct subgroup within Lineage 1a. In contrast, Lineage 2 consists of WNV strains from mosquitoes (Culex pipiens) in Iran (2017 and 2018), which is also supported by high bootstrap values (100%) as seen, highlighting the geographical and host diversity of WNV in the MENA region.
An expanded phylogenetic analysis was constructed with partial and complete WNV sequences using the Neighbor-Joining (NJ) method (Figure 2). The study yielded high bootstrap values (85% to 100%) across key nodes, indicating strong confidence in grouping sequences. The results show that while complete sequences cluster into two primary lineages (Lineage 1a and Lineage 2), the partial sequences exhibit a more diverse branching pattern based on host species and geographical origin. Most of the sequences in the tree are from Israel, with additional sequences from the UAE, Morocco, Tunisia, Iran, Iraq, and Egypt, providing a comprehensive view of WNV diversity across the MENA countries. Lineage 1a consists of sequences from Israel (2000-2010) and Tunisia (2003), with a recent addition of the Iraq 2023 strain (PP430512.1, from Anatidae), which clusters closely with older sequences, suggesting the continued circulation of Lineage 1a in the region.
In contrast, Lineage 2 includes sequences from Israel and Iran with key strains from 2004 (Israel Sarafend) and more recent isolates from Culex mosquitoes in Iran (2017-2018). Generally, the dataset represented includes a variety of hosts, such as humans, birds (Ciconiidae), and mammals (e.g., Camelus dromedarius). The mosquito-derived sequences dominate the dataset as primary vectors for WNV, especially for Lineage 2. Notably, the close relationship between the Iraq 2023 strain and other human/animal-derived isolates within Lineage 1a demonstrates the virus’s ability to infect several hosts with potential cross-species and -lineage transmission. This was essential for understanding the virus’s epidemiology in the region; thus, further analysis was conducted to compare the partial sequences associated with each lineage.

We constructed a Neighbor-Joining (NJ) phylogeny with complete sequences of Lineage 1a and Lineage 2 as references for insight into their evolutionary relationships with partial sequences (Figure 3). Lineage 1a is represented by AF481864.1 (2002, Israel, Ciconiidae), while Lineage 2 is represented by AY688948.1 (2004, Israel, Sarafend). Both reveal distinct relations to sequences in humans and mosquitoes from Israel (2000, 2009, 2010) and Iraq (2023). The tree also shows diversity in the cluster pattern for partial sequences, where some align more closely with Lineage 1a and others with Lineage 2, while mosquitoes are the dominant vectors of the ongoing circulation. The Iraq 2023 strain (PP430512.1) is closely related to Lineage 1a, particularly the older strain AF481864.1 (2002 Israel Ciconiidae). Its position within Lineage 1a and the evolutionary linkage between avian and human-derived strains from Israel show that these lineages have continued to evolve and transmit within the region, including Iraq.

Similar to the previous analysis but represented differently, we used AlignStatPlot to represent multiple sequence alignment (MSA) plots and show the standard nucleotides between WNV sequences to study genetic diversity. In Figure 4, we analyzed 26 sequences generated using AlignStatPlot, a web-based tool for sequence alignment statistics and visualization. The sequences range from 1,100 to 11,700 base pairs (bp), and the plot illustrates genetic similarities and differences among the WNV strains. Furthermore, the inner rings represent older or more conserved sequences, and the outer rings indicate newer or more recently diverged ones, highlighting the evolutionary dynamics of WNV across different periods and geographic locations. They display a transparent region of high similarity, particularly in the conserved regions of the genome; these regions are often associated with essential viral functions and exhibit slight genetic variation. Sequences closer to each other within the same segment of a ring share a high level of gene similarity. These findings underscore the intricate relationships among WNV strains and provide insight into how genetic variations might influence transmission dynamics and disease emergence.

Further analysis focused on five major viral proteins, NS1, NS2, NS3, NS4, and NS5, due to their roles in the virus's replication, immune evasion, and virulence. We used the sequences of AF481864.1 (2002 Israel Ciconiidae, Lineage 1a), AY268133.1 (2003 Tunisia HS, Lineage 1a), AY688948.1 (2004 Israel Sarafend, Lineage 2), and MN238670.1 (2018 Iran Culex theileri) in the comparison—the selection criteria were explained in the methods section. Supplementary Table 1 (A–E) shows the amino acid sites with their respective differences and similarities at the same positions across each sequence; the results reveal several amino acid substitutions across all proteins, with significant variation in positions that impact viral fitness and host interactions. Notably, the 2004 Israel Sarafend (Lineage 2) and the 2018 Iran Culex theileri strains showed high divergence compared to the older Lineage 1a strains (AF481864.1 and AY268133.1). We classified the severity of the amino acid changes based on the type of alteration: if the change was to the same polarity or charge, it was classified as low. They were considered minor if the charge remained unchanged but significantly differed inside chain size. If the change was in polarity, it was classified as either moderate or minimal, depending on the extent of the side chain change. However, alterations involving a charge only (or with polarity and size) were classified as severe, as these can profoundly impact the protein’s three-dimensional structure by disrupting the residue's interactions and stability, potentially leading to protein dysfunction or disease [22-24].
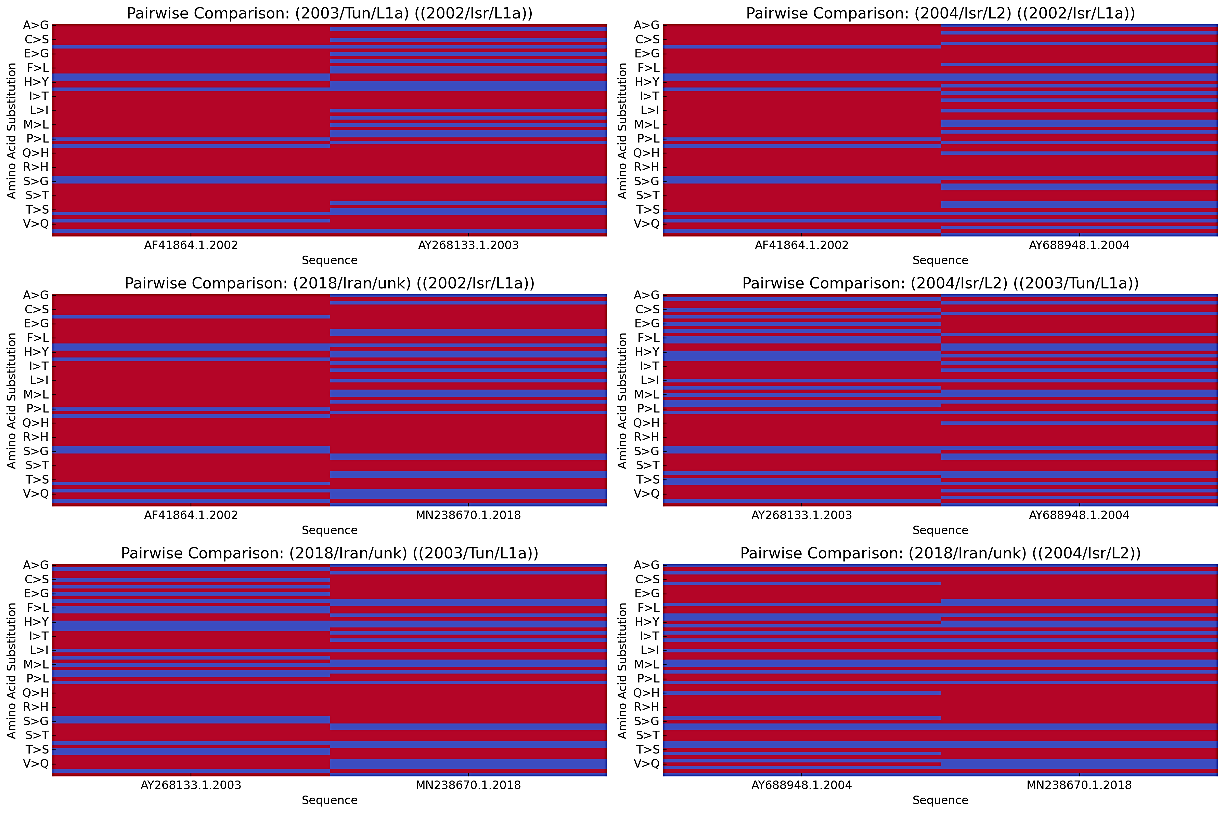
A high similarity was observed between the Iraq 2018 strain and the Lineage 2 sequence compared to the Israel 2002 reference sequence, with critical substitutions identified at the following positions: NS1 (K874A, E917N, K964E, R1028G, Q1172R); NS2 (H1297Y, L1301S, R1340G, G1488D, E1489G); NS3 (Q1629H, P1786H, T1973E); NS4 (Q2324H); and NS5 (Q2579H, K2700A, R2758H, N2841, E2859V, E2859A, H3011N, G3366K), all of severe impact which indicates a potential changes in viral function that may affect its pathogenicity or immune evasion. A Venn diagram was also made (Figure 5) to highlight the strains' shared and unique amino acid positions. Among three WNV sequences, Israel 2002 (AF481864.1), Tunisia 2003 (AY268133.1), and Iran 2018 (MN238670.1). Among these, 10 were shared between (AF481864.1) and (AY268133.1). 14 positions were unique to (AF481864.1), 2 were unique to (AY268133.1), and 44 were unique to (MN238670.1). The 2018 Iran strain exhibited the highest unique amino acid positions, suggesting it has undergone more recent evolutionary changes. As it shows, the similarities in amino acid sequences outweigh the differences, indicating a high degree of conservation at the protein level. Next, a pairwise comparison of amino acid substitutions was performed using R programming between the current sequences, analyzing two at a time (Figure 6); the X-axis represents the specific viral sequences being compared, and the Y-axis displays the amino acid substitutions observed between the sequences, with each letter pair indicating the original and substituted amino acids. Additionally, the colors illustrate the level of similarity or divergence between the amino acid sequences being compared. The red bars indicate identical regions, meaning high similarity (no substitutions) at those positions, while the blue bars correspond to areas of amino acid substitution. This effectively differentiates conserved regions (red) and evolutionary change areas (blue). Thus, the Iran strain 2018 (MN238670.1) appears to be more closely related to Lineage 1a and shows distinct differences from both lineages.

.
Our study provided insights into the phylogenetic relationships, evolutionary patterns, and the ability of WNV to persist across various hosts and geographies in the MENA region. Some studies hint at the reemergence of WNV in Europe and America; however, to our knowledge, this is the first complete phylogenetic study of WNV in the MENA region in 2024. We found that the only published sequence close to our research was the strain isolated in Iraq in 2023; as a partial sequence, it is very closely related to Lineage 1a—particularly with the Israel 2002 strain (AF481864.1). The latest complete sequence in the MENA before publishing our analysis was from Iran in 2018, which has a closer phylogenetic relationship with Lineage 2. These results confirm the presence of lineages 1a and 2 and indicate a high genetic diversity in these lineages, mainly among viral sequences from Israel, Iraq, and Iran. Such variations are evident throughout the many substitutions found in the non-structural proteins NS1, NS2, NS3, NS4, and NS5, which are involved in viral replication, immune evasion, and host interactions [25]. Analyzing amino acid substitution patterns across the significant viral proteins (NS1-NS5) adds another layer of complexity to understanding how WNV adapts and evolves. As seen in Supplementary Table 1, the NS5 protein had the most notable substitutions, which are likely to impact the virus's RNA-dependent RNA polymerase (RdRp) function and methyltransferase activity, as it contains both an N-terminal methyltransferase (MTase) and a C-terminal RdRp domain, which are essential for viral RNA replication and immune evasion; making this protein a primary target for host immune responses [26].
Furthermore, substitutions in NS5—especially those that alter key functional domains—may enhance the virus's capacity to evade immune detection or increase its replication efficiency, thereby contributing to viral persistence and virulence; such mutations are known to occur in response to selective pressures from the host immune system, with significant effects on viral replication and immune evasion [27, 28]. The accumulation of mutations in NS5, combined with changes in other proteins like NS1 and NS3, reflects the virus's broader strategies for adapting to host environments and overcoming immune pressures. Moreover, identifying unique amino acid changes in the 2018 Iran strain compared to older Lineage 1a strains suggests that Lineage 2 may undergo more rapid evolutionary changes, which could influence its epidemiological behavior in the region. In this arboviral nature, mosquitoes are mostly recognized as the principal vectors transmitting the virus to humans and other hosts, which calls for additional observation and careful consideration.
From our review, recorded infections in humans and animals suggest a route for zoonotic transmission. It is also worth pointing out that, among the various countries, sequences from Israel are the most abundant, dating from 2000 to 2018. It has been confirmed by recent reports of West Nile disease in 2024 that WNV is actively spreading among Israeli mosquito populations and other hosts, such as humans and birds [29]. The predominance of Israeli sequences suggests that the virus is either sampled more intensely in this country or circulated more widely within it than in other parts of the MENA region. However, this does not mean that WNV is localized to Israel by any means; it does place it at the center of attention regarding WNV research and surveillance. Other papers have suggested that climate and environmental factors, including rising temperatures, could have promoted genetic variation and increased mutation rates of WNV [30]; such phenomena may account for the substantial difference in the variable regions between older reference sequences, such as the Israel 2002 strain and new strains like the Iraq 2023 and Iran 2018 strain. This fact alone supports the need for constant monitoring and analysis of WNV in the region due to potential climate-induced modifications.
The findings indicate that the virus is circulating in the region based on newly available database sequences. Still, unfortunately, not all were of sufficient quality to be included in our study. This does not imply the absence of WNV cases in other countries within the region; instead, it points to underreporting or lack of sequencing efforts; the fact that WNV cases have been reported in countries like Jordan, Syria, Sudan, and Pakistan [2], but without extensive sequencing data, points to a broader issue of underreporting and limited genomic surveillance. While these countries may not have as many sequenced cases, the presence of WNV-related instances indicates that the virus is circulating beyond the countries where sequencing data is available, such as Israel and Iran. This suggests that the extent of WNV in the MENA region may be underestimated due to gaps in sequencing and reporting. Improving surveillance infrastructure and encouraging more widespread sequencing efforts across the region will be essential for obtaining a more accurate understanding of WNV’s transmission patterns and evolutionary changes [31]. Currently, WNV has been demonstrated to be circulating again in Europe, heightening concerns about its potential spread northward [31], focusing on the potential for WNV outbreaks in previously underreported or unreported countries, and reinforcing the importance of regional cooperation and comprehensive vector control strategies.
West Nile Virus (WNV) poses a significant public health risk due to its ability to infect humans and animals. This is why effective biosafety and biorisk management are critical for controlling the spread of WNV in the MENA region. Environmental conditions, including rising temperatures and changing rainfall patterns, create ideal habitats for Culex mosquitoes, the primary vectors of WNV [32]. The continued evolution of the virus—as seen in the genetic differences between strains such as Iraq 2023 and Iran 2018—raises concerns about its adaptability and spreading to previously unaffected areas, leading to potential public health crises. The possibility of preventing WNV transmission and future outbreaks underscores the importance of regional cooperation in enhancing surveillance systems and implementing effective response strategies, with each country adjusting them based on its epidemiological status and target hosts [33]. Given the zoonotic potential of WNV, stringent biosafety protocols are needed in both research and diagnostic laboratories. Handling viral samples from infected animals, humans, and mosquitoes requires containment measures such as BSL-3 (Biosafety Level 3) environments, where appropriate personal protective equipment (PPE) and engineering controls are in place to prevent accidental exposure or release of the virus, with regular training of laboratory personnel which is essential to ensure adherence to safety protocols making the region ready for any viral attacks in the future [8, 34]. Likewise, controlling the mosquito population remains one of the most effective methods for preventing WNV transmission; for example, vector control programs include insecticide applications, the removal of mosquito breeding sites, and public awareness campaigns on personal protection measures such as repellents and mosquito nets. All these efforts must be coordinated regionally, as WNV can spread across borders, requiring a collaborative approach to monitoring and response strategies [8, 35]. Vaccines are an excellent and promising approach to prevent WNV circulation, especially for end hosts like humans or horses. However, there is currently no approved vaccine for humans; effective veterinary vaccines are available, which could be a significant step toward controlling and potentially eradicating flavivirus infections [36].
This study sheds light on the current state of WNV in the MENA region and raises essential questions about the virus’s potential for further spread and adaptation. The findings underscore the importance of ongoing genomic surveillance, vector control efforts, and public health preparedness to mitigate the risk of future outbreaks. As WNV continues circulating and evolving, countries must invest in enhanced sequencing capabilities and collaborate on regional efforts to monitor and control this persistent and adaptable virus. Managing the biosafety and biorisk of WNV in the MENA region requires a multifaceted approach that integrates enhanced surveillance, reinforced laboratory safety, effective vector control, and regional cooperation. As the virus evolves and adapts, proactive measures will be essential in mitigating the risk of future outbreaks and protecting public health, especially in developing countries.
1. Liang Y, Dai X. The global incidence and trends of three common flavivirus infections (Dengue, yellow fever, and Zika) from 2011 to 2021. Front Microbiol. 2024;15:1458166. https://doi.org/10.3389/fmicb.2024.1458166
2. Eybpoosh S, Fazlalipour M, Baniasadi V, Pouriayevali MH, Sadeghi F, Ahmadi Vasmehjani A, et al. Epidemiology of West Nile Virus in the Eastern Mediterranean region: A systematic review. PLoS Negl Trop Dis. 2019;13(1):e0007081. https://doi.org/10.1371/journal.pntd.0007081
3. Fereidouni S. Ecology of Wild Bird Diseases. In: Fereidouni S, Hawksworth DL, Samson RA, editors. Ecology of Wild Bird Diseases. CRC Press; 2024. p. 1–11.
4. Chancey C, Grinev A, Volkova E, Rios M. The global ecology and epidemiology of West Nile virus. Biomed Res Int. 2015;2015:376230. https://doi.org/10.1155/2015/376230
5. Sejvar, J.J., West nile virus: an historical overview. Ochsner J, 2003. 5(3): p. 6-10. Accessed: March 28, 2025. Available from: https://www.ochsnerjournal.org/content/5/3/6
6. Sayed-Ahmed M. Incidence History of West Nile Virus in Africa and Middle East, With an Emphasis on Egypt: A Review. J Dairy Vet Anim Res. 2016;3(3):77–82. Accessed March 28, 2025. Available from: https://medcraveonline.com/JDVAR/incidence-history-of-west-nile-virus-in-africa-and-middle-east-with-an-emphasis-on-egypt-a-review.html https://doi.org/10.15406/jdvar.2016.03.00080
7. García-Carrasco, J.M., et al., Mapping the Risk for West Nile Virus Transmission, Africa. Emerging Infectious Diseases, 2022. 28(4): p. 777-785. https://doi.org/10.3201/eid2804.211103
8. Centers for Disease Control and Prevention (CDC). Guidelines for West Nile Virus Surveillance and Control | West Nile Virus | CDC. Accessed March 28, 2025. Available from: https://www.cdc.gov/west-nile-virus/php/surveillance-and-control-guidelines/index.html
9. Petersen LR, Brault AC, Nasci RS. West Nile virus: review of the literature. JAMA. 2013;310(3):308–15. https://doi.org/10.1001/jama.2013.8042
10. Sichangi MW. Current state of affairs for West Nile Virus: Review. Int J Mosq Res. 2021;8(4):10–15. Accessed March 28, 2025. Available from: https://ijiset.com/vol8/v8s4/IJISET_V8_I04_21.pdf
11. Fang Y, Khater EIM, Xue JB, Ghallab EHS, Li YY, Jiang TG, et al. Epidemiology of Mosquito-Borne Viruses in Egypt: A Systematic Review. Viruses. 2022;14(7):1502. https://doi.org/10.3390/v14071577
12. Paz S. The West Nile Virus outbreak in Israel (2000) from a new perspective: the regional impact of climate change. Int J Environ Health Res. 2006;16(1):1–13. https://doi.org/10.1080/09603120500392400
13. Schaffner F, Bansal D, Mardini K, Al-Marri SA, Al-Thani MHJ, Al-Romaihi H, et al. Vectors and vector-borne diseases in Qatar: current status, key challenges and future prospects. J Eur Mosq Control Assoc. 2021;39(1):3–13. https://doi.org/10.52004/JEMCA2021.x001
14. Murgue B, Murri S, Triki H, Deubel V, Zeller HG. West Nile in the Mediterranean Basin: 1950–2000. Ann N Y Acad Sci. 2001;951:117–26. https://doi.org/10.1111/j.1749-6632.2001.tb02690.x
15. Lourenco J, Thompson RN, Theze J, Obolski U. Characterising West Nile virus epidemiology in Israel using a transmission suitability index. Euro Surveill. 2020;25(46):1900629. https://doi.org/10.2807/1560-7917.ES.2020.25.46.1900629
16. Al-Tayib OA. An Overview of the Most Significant Zoonotic Viral Pathogens Transmitted from Animal to Human in Saudi Arabia. Pathogens. 2019;8(1):25. https://doi.org/10.3390/pathogens8010025
17. Alzuheir I, Fayyad A, Jalboush N, Abdallah R, Abutarbush S, Gharaibeh M, et al. Seroprevalence and risk factors of West Nile virus infection in veterinarians and horses in Northern Palestine. Vet World. 2021;14(5):1241–6. https://doi.org/10.14202/vetworld.2021.1241-1246
18. Imran M, Ye J, Saleemi MK, Shaheen I, Zohaib A, Chen Z, et al. Epidemiological trends of mosquito-borne viral diseases in Pakistan. Anim Dis. 2022;2(1):4. https://doi.org/10.1186/s44149-021-00034-4
19. Alsamman AM, El Allali A, Mokhtar MM, Al-Sham’Aa K, Nassar AE, Mousa KH, et al. AlignStatPlot: An R package and online tool for robust sequence alignment statistics and innovative visualization of big data. PLoS ONE. 2023;18(9):e0291204. https://doi.org/10.1371/journal.pone.0291204
20. Kinene, T., et al., Rooting Trees, Methods for. 2016, Elsevier. p. 489-493.
21. Hess, P.N. and C.A. De Moraes Russo, An empirical test of the midpoint rooting method. Biological Journal of the Linnean Society, 2007. 92(4): p. 669-674. https://doi.org/10.1111/j.1095-8312.2007.00864.x
22. Costa D, Pradier C-M, Tielens F, Savio L. Adsorption and self-assembly of bio-organic molecules at model surfaces: A route towards increased complexity. Surf Sci Rep. 2015;70(4):449–553. https://doi.org/10.1016/j.surfrep.2015.10.002
23. Rehman I, Kerndt CC, Botelho S. Biochemistry, Tertiary Protein Structure. In: StatPearls (Internet). Treasure Island (FL): StatPearls Publishing; 2024. Accessed March 28, 2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470269/
24. Teng S, Srivastava AK, Schwartz CE, Alexov E, Wang L. Structural assessment of the effects of amino acid substitutions on protein stability and protein–protein interaction. Int J Comput Biol Drug Des. 2010;3(4):334–49. https://doi.org/10.1504/IJCBDD.2010.038396
25. Geiss BJ, Stahla H, Hannah AM, Gari HH, Keenan SM. Focus on Flaviviruses: Current and Future Drug Targets. Future Med Chem. 2009;1(2):327–44. https://doi.org/10.4155/fmc.09.27
26. Li X-D, Shan C, Deng C-L, Ye H-Q, Shi P-Y, Yuan Z-M, et al. The Interface between Methyltransferase and Polymerase of NS5 Is Essential for Flavivirus Replication. PLoS Negl Trop Dis. 2014;8(5):e2891. https://doi.org/10.1371/journal.pntd.0002891
27. Caldwell HS, Lasek-Nesselquist E, Follano P, Kramer LD, Ciota AT. Divergent Mutational Landscapes of Consensus and Minority Genotypes of West Nile Virus Demonstrate Host and Gene-Specific Evolutionary Pressures. Genes. 2020;11(11):1299. https://doi.org/10.3390/genes11111299
28. Youn S, Ambrose RL, Mackenzie JM, Diamond MS. Non-structural protein-1 is required for West Nile virus replication complex formation and viral RNA synthesis. Virol J. 2013;10:339. https://doi.org/10.1186/1743-422X-10-339
29. Mor Z, Omari H, Indenbaum V, Kirstein OD, Shatach Catabi O, Reicher S, et al. Early rise of West Nile fever in Israel, June 2024. Euro Surveill. 2024;29(30):2400457. https://doi.org/10.2807/1560-7917.ES.2024.29.30.2400457
30. Fay RL, Ngo KA, Kuo L, Willsey GG, Kramer LD, Ciota AT. Experimental Evolution of West Nile Virus at Higher Temperatures Facilitates Broad Adaptation and Increased Genetic Diversity. Viruses. 2021;13(10):1889. https://doi.org/10.3390/v13101889
31. Lu L, Zhang F, Oude Munnink BB, Munger E, Sikkema RS, Pappa S, et al. West Nile virus spread in Europe: Phylogeographic pattern analysis and key drivers. PLoS Pathog. 2024;20(1):e1011880. https://doi.org/10.1371/journal.ppat.1011880
32. Wang HR, Liu T, Gao X, Wang HB, Xiao JH. Impact of climate change on the global circulation of West Nile virus and adaptation responses: a scoping review. Infect Dis Poverty. 2024;13(1):38. https://doi.org/10.1186/s40249-024-01207-2
33. Young JJ, Coulombier D, Domanović D, Zeller H, Gossner CM. One Health approach for West Nile virus surveillance in the European Union: relevance of equine data for blood safety. Euro Surveill. 2019;24(16):1800349. https://doi.org/10.2807/1560-7917.ES.2019.24.16.1800349
34. McKellar DR, Conway MJ. Safe Handling of West Nile Virus in the Insectary. In: West Nile Virus. Methods Mol Biol. 2016;143–50.
35. Bellini R, Zeller H, Van Bortel W. A review of the vector management methods to prevent and control outbreaks of West Nile virus infection and the challenge for Europe. Parasites Vectors. 2014;7:323. https://doi.org/10.1186/1756-3305-7-323
36. Habarugira G, Suen WW, Hobson-Peters J, Hall RA, Bielefeldt-Ohmann H. West Nile Virus: An Update on Pathobiology, Epidemiology, Diagnostics, Control and “One Health” Implications. Pathogens. 2020;9(7):589. https://doi.org/10.3390/pathogens9070589
37. VectorSurv. Vectorborne Disease Surveillance System. [Accessed March 28, 2025]. Available from: https://vectorsurv.org/